Sequencing the microbiome
The sequencing of the human genome was a giant undertaking by itself. However, the scientists working on the HMP had perhaps an even more daunting task at hand: the human microbiome consists of thousands of different species of microorganisms, each with their own unique genome. Adding an additional challenge is the fact that many of these microbes are unable to be grown by themselves in a laboratory setting. To tackle these challenges, researchers used two main methods of DNA sequencing to analyze their samples. The two different methods are explained in the following sections.
16S rRNA gene sequencing
Our ability to describe complex microbial communities has been limited by the fact that many organisms within these communities remain unculturable. In order to classify the microorganisms that make up the human microbiome, scientists used 16S rRNA gene sequencing. The 16S rRNA is a component of the 30S subunit of prokaryotic ribosomes. The gene encoding the 16S rRNA is often used in phylogenetic studies in order to identify bacteria present in a community and to map their relationship to each other.
Our ability to describe complex microbial communities has been limited by the fact that many organisms within these communities remain unculturable. In order to classify the microorganisms that make up the human microbiome, scientists used 16S rRNA gene sequencing. The 16S rRNA is a component of the 30S subunit of prokaryotic ribosomes. The gene encoding the 16S rRNA is often used in phylogenetic studies in order to identify bacteria present in a community and to map their relationship to each other.

Figure 1: An example of a 16S rRNA gene. The regions in green are conserved in all microorganisms. These are the sites that are targeted by primers for PCR amplification so that all the 16S rRNA genes in a sample are amplified. The grey regions are the species-specific regions that-- when sequenced-- allow for scientists to see which species are present in a community.
Image courtesy of: http://www.alimetrics.net/en/index.php/dna-sequence-analysis
The 16S rRNA gene is used because it has regions that are highly conserved among all prokaryotes (Figure 1). This means that these regions of the gene are unchanged from species to species and are found in almost all prokaryotes. This conservation makes it easy for scientists to design universal primers to target the 16S rRNA gene for PCR amplification in all microorganisms. When DNA that has been extracted from a community of microorganisms is PCR amplified with these primers, all the 16S rRNA genes from all the species present are amplified. However, other regions of the gene are species-specific. This allows scientists to identify which species are present in a community once the gene has been sequenced. (See Figure 2 for more specific details on the sequencing process) .
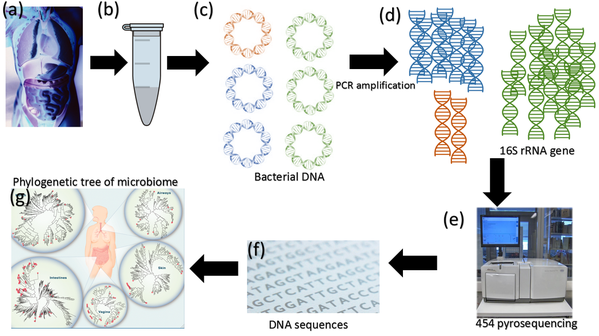
Figure 2. (a) and (b) Researchers take samples from various sites of the human body. (c) Prokaryotic DNA is extracted from the samples. The composition of the DNA is dependent upon which species are present and how abundant each individual species is. (d) PCR is used to amplify the 16S rRNA gene from all the different prokaryotic chromosomes. PCR allows scientists to generate many copies of a gene from only a few copies. Having many copies makes it much easier to sequence a gene. The amount of copies that are present is approximately proportional to the abundance of each species in the original sample. (e) A sequencing technique called "454 pyrosequencing is used to generate (f) sequences for each of the 16S rRNA genes amplified. (g) Researchers then compile this data into a phylogenetic tree.
Metagenomic sequencing
While 16S rRNA gene sequencing provides a good estimate of who is there, this data does not tell scientists about what the microbes could be doing inside our bodies. In order to get a better idea of how these microbes live their lives inside our bodies, scientists had to turn to metagenomic sequencing. Metagenomics is the study of the total DNA extracted from an environmental (in this case the human body!) sample. Instead of having to culture a specific microorganism, extract its DNA, and then sequence the DNA, metagenomic sequencing allows scientists to directly extract and sequence DNA from their environmental sample.
Scientists typically use a method called "shotgun sequencing" in order to sequence the metagenome of the human microbiome (Figure 2). In shotgun sequencing, all the pieces of DNA within a sample are cut up into very small pieces. The DNA has to be cut up into small pieces because the machines that are used currently to sequence DNA are not yet good enough to sequence very long pieces of DNA. After the DNA is cut up into the small pieces. Each of the small pieces is then sequenced. Since the resulting sequences contain small regions that overlap with other sequences, computer programs can then be used to piece together these sequences as best as possible. It is important to note that it is often not possible to recreate the entire genome of all the organisms present in a sample. Instead, scientists are left with multiple fragments of the different genomes. These fragments can then be compared to the complete genomes of other, known organisms as well as various databases of genetic information in order to figure out which bacteria were originally present in the sample as well as what genes they have in their genomes. The information about what genes are present helps us to predict how these organisms might be functioning within the human body.
While 16S rRNA gene sequencing provides a good estimate of who is there, this data does not tell scientists about what the microbes could be doing inside our bodies. In order to get a better idea of how these microbes live their lives inside our bodies, scientists had to turn to metagenomic sequencing. Metagenomics is the study of the total DNA extracted from an environmental (in this case the human body!) sample. Instead of having to culture a specific microorganism, extract its DNA, and then sequence the DNA, metagenomic sequencing allows scientists to directly extract and sequence DNA from their environmental sample.
Scientists typically use a method called "shotgun sequencing" in order to sequence the metagenome of the human microbiome (Figure 2). In shotgun sequencing, all the pieces of DNA within a sample are cut up into very small pieces. The DNA has to be cut up into small pieces because the machines that are used currently to sequence DNA are not yet good enough to sequence very long pieces of DNA. After the DNA is cut up into the small pieces. Each of the small pieces is then sequenced. Since the resulting sequences contain small regions that overlap with other sequences, computer programs can then be used to piece together these sequences as best as possible. It is important to note that it is often not possible to recreate the entire genome of all the organisms present in a sample. Instead, scientists are left with multiple fragments of the different genomes. These fragments can then be compared to the complete genomes of other, known organisms as well as various databases of genetic information in order to figure out which bacteria were originally present in the sample as well as what genes they have in their genomes. The information about what genes are present helps us to predict how these organisms might be functioning within the human body.
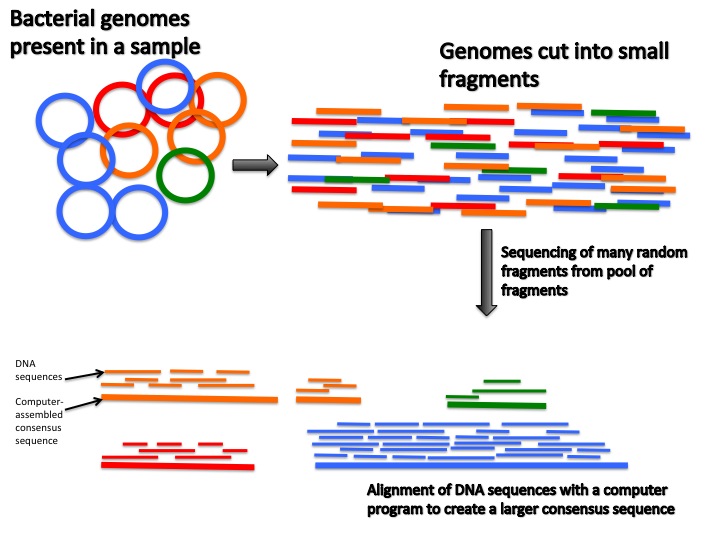
Figure 3. For metagenomic sequencing, bacterial genomes are extracted from a sample. Since the genomes are very large, they have to be cut into much smaller fragments that are more easily sequenced. The small fragments are then randomly sequenced many many times in order to sequence as many of the fragments as possible. The reason why the sequencing needs to be done a large amount of times is that the abundance of each genome in the sample will vary depending on the relative abundance of the bacteria it came from. If you only randomly sequenced your sample of fragments a few times, then only the most common genome fragments would end up being sequenced and the genome fragments from the less abundant bacteria would never be seen.
The DNA sequences that are produced from the sequencing have ends that overlap with each other. A computer program then fits all these sequences together like pieces of a puzzle in a process called "alignment". Once the alignment is complete, the program then generates what is called a "consensus sequence" which is representative of the original DNA sequences in the bacterial genome. Unfortunately, due to limits in sequencing technology it is often not possible to sequence the entire genome of every single bacterial species in a sample. Instead, the more abundant bacteria end up having the more complete genomes due to being more represented in the sequences and the less abundant bacteria have much more incomplete genomes.
